Opinion Article - (2022) Volume 10, Issue 4
COMPUTER-AIDED ENGINEERING OF ENHANCING ANTIBODIESâ IMMUNOGENICITY
Hyun Mi-wol*Received: Nov 01, 2022, Manuscript No. JHRE-22-85083; Editor assigned: Nov 04, 2022, Pre QC No. JHRE-22-85083 (PQ); Reviewed: Nov 18, 2022, QC No. JHRE-22-85083; Revised: Nov 25, 2022, Manuscript No. JHRE-22-85083 (R); Published: Dec 05, 2022, DOI: 10.30876/2347-7393.22.10.211
Description
Computational techniques have received a lot of interest recently in protein engineering. To evaluate the sequences and structures of proteins and to predict their varied features, wide ranges of computational techniques have been developed. Since antibodies are one of the emerging protein therapies, it is extremely desirable to find ways to regulate their physicochemical characteristics. Nevertheless, despite the enormous work done over the past few decades, computational methods to predict the physicochemical features of antibodies are still in their infancy. For real-world applications, experimental validations are unquestionably necessary, and the findings should be handled with care. Therefore, in analysis they concentrate on the stability, viscosity, and immunogenicity of antibodies, as well as the state of computational techniques for modifying these properties.
By assisting with activities like screening potential compounds, determining drug similarity, and optimising physicochemical and pharmacokinetic features, computational approaches have become crucial tools for both drug discovery and antibody engineering. To examine the quantitative structure-activity connections of target molecules in various situations, a wide variety of computational techniques have been developed. Particularly in antibody engineering, the increase in antibody crystal structures allowed us to investigate the links between sequence and structure, which resulted in the development of techniques for high-resolution antibody modelling. Various features of antibodies have been predicted using computational methods based on sequences or structures. However, those techniques are still in their infancy despite the enormous efforts of previous decades. For real- world applications, experimental validations are unquestionably necessary, and the findings should be handled with care.
In many different fields, antibodies are crucial molecules. A number of antibody qualities need to be designed depending on their intended uses. The most significant physicochemical characteristic may be binding affinity, which is a quantitative indicator of how antibodies naturally perform. Six complementarity-determining areas on antibodies allow them to attach to foreign molecules or antigens (CDRs). Despite the limited conformations, that 5 of the CDRs (L1, L2, L3, H1, and H2) assume the significant diversity of CDR-H3 in terms of sequence and structure It can identify an endless number of antigens. Affinity maturation engineering has focused on modifying the CDRs’ amino acid sequences since antibodies work through their six CDRs. It is well recognised that antibody engineering has a problem with the trade-off between binding affinity and other qualities. Improving other qualities like as stability, viscosity, and immunogenicity can therefore be realised by modifying the amino acids of the non-CDR sections of antibodies. To produce better antibodies, careful evaluation of mutational sites is frequently required. Random mutagenesis is one of the most often used techniques for enhancing antibody characteristics to get over such tiresome processes. Not just binding affinities but also other qualities can be engineered. By raising the temperature and managing the parameters of the solution during the selection process. However, computational design is evolving into a different approach in antibody engineering because of recent improvements in computational power and algorithms. One benefit of computational approaches is that, when used in conjunction with a structure, their use can be rational. Physical laws should control antibodies and their structures, and based on these laws, we should be able to predict how antibodies will behave in solutions and within the human body. The accuracy of computational algorithms is inferior to that of library-based methods due to our inadequate grasp of the biophysical principles of biomolecules and the difficulty in characterising conformational dynamics; therefore, such predictions are not very satisfactory. Present a current assessment of computational designs for the physicochemical aspects of antibodies, which have not been discussed in earlier reviews on computer-aided antibody design. In computer-aided antibody design, particular attention has been paid to the stability, viscosity, and immunogenicity of antibodies, among other features.

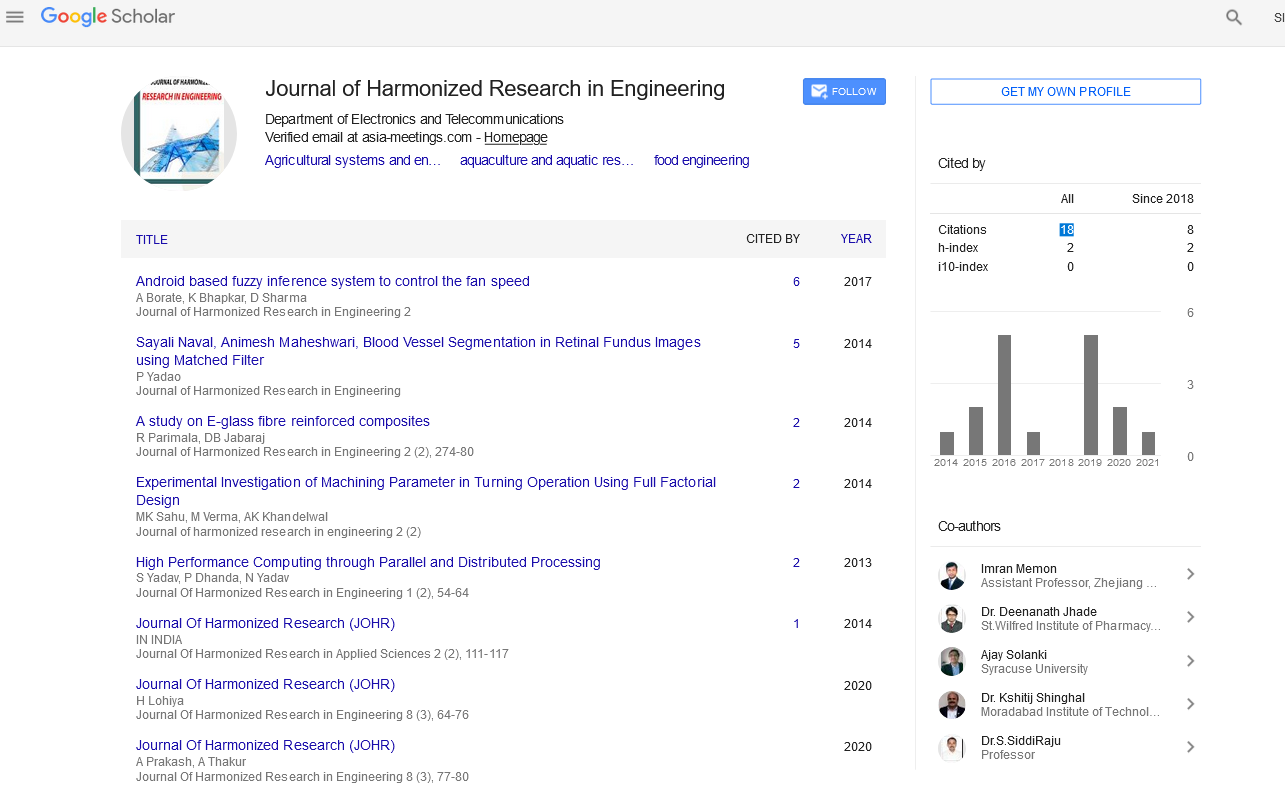
Google Scholar citation report
Citations : 43
Journal of Harmonized Research in Engineering received 43 citations as per google scholar report